This workflow demonstrates a simple (and pretty standard) flexible molecular alignment workflow: diverse conformers are generated for a set of database molecules which are then aligned to a query molecule. The highest scoring conformer for each database molecule is kept.

Conformer generation is done using the RDKit distance geometry implementation. The resulting conformers are cleaned up with the MMFF94 force field and then an RMS filtering step is applied to remove redundant conformers. The meat of the workflow is mostly hidden in the metanodes, so I set up some quickforms to make configuration easy. Here's the configure dialog for the "Generate Diverse Conformers" metanode:
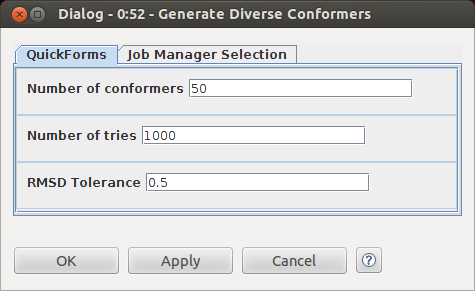
Neither the RDKit nor Knime itself provide a 3D molecule visualizer, so you'll need to install something else to see the results. Here I used the Interactive BioSolveIT Viewer, which is freely available from their website, there's also a link in the workflow. Here are a couple of the alignments that were generated, the query molecule is in green and the database molecule is gray.


You can see that decent conformers are being generated and that the alignments are quite good.
Until I figure out how to host these in a sensible way on a public knime server, I'll just provide things via dropbox links.
Here's the data directory that is used in this and all other workflows: https://db.tt/qSdJn0St
And here's the link to this workflow: https://db.tt/4WOxcXWi
No comments:
Post a Comment